Overview
Huntington’s disease (HD) is a rare, inherited disease that causes the progressive breakdown (degeneration) of nerve cells in the brain. HD has a broad impact on a person’s functional abilities and usually results in movement, thinking (cognitive) and psychiatric symptoms.
HD is known as a family disease because every child of a parent with HD has a 50/50 chance of inheriting the faulty gene. Today, there are approximately 41,000 symptomatic Americans and more than 200,000 at-risk of inheriting the disease.
The mutation, causing HD, is found in the gene for huntingtin which was discovered in 1993. HD is caused by a stretch of nucleotide C-A-G which is repeated abnormally, known as the CAG repeat expansion. In the huntingtin gene, most healthy individuals have around 20 CAG repeats, however people with HD have 36 or more. Every person who has this CAG repeat expansion in the HD gene will eventually develop the disease, and each of their children have a 50% chance of developing HD.
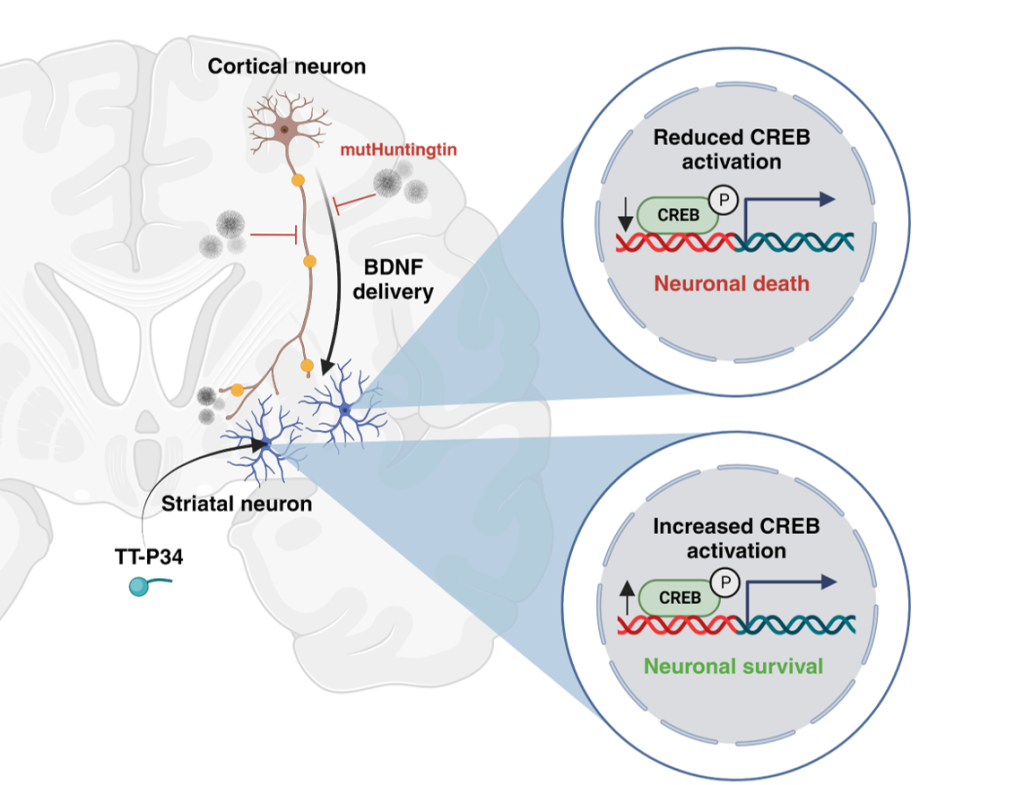
The mutated Huntingtin protein (mHTT) aggregates and thereby prevents key functions in the cell – ultimately leading loss of neurotrophic support and lysosomal and mitochondrial dysfunction. This leads to loss of neurons and development of symptoms of HD.
Our peptide is able to re-activate the functions that are inhibited, thereby rebalancing the neuronal function, not only ensuring slowing of disease (removal of new mHTT) but also disease modification by ensuring survival and function of neurons.